Damietta protein design engine
The core of the Damietta Toolkit is a physics-based design engine that combines the advantages of speed, generalizability and self-consistent scoring.
Accelerated energy calculations
Computational protein design involves evaluating the energy for numerous combinations of amino acid identities and side chain conformations. Traditionally, the non-bonded interaction energy between an inbound rotamer and its molecular environment is calculated through exhaustive looping over all interacting pairs of atoms within a distance cutoff. In contrast, Damietta framework substitutes these calculations with a single operation between two large matrices (tensors) representing two groups of atoms. This considerably reduces runtime load and allows to achieve ideal process parallelization, due to uniform tensor dimensions.
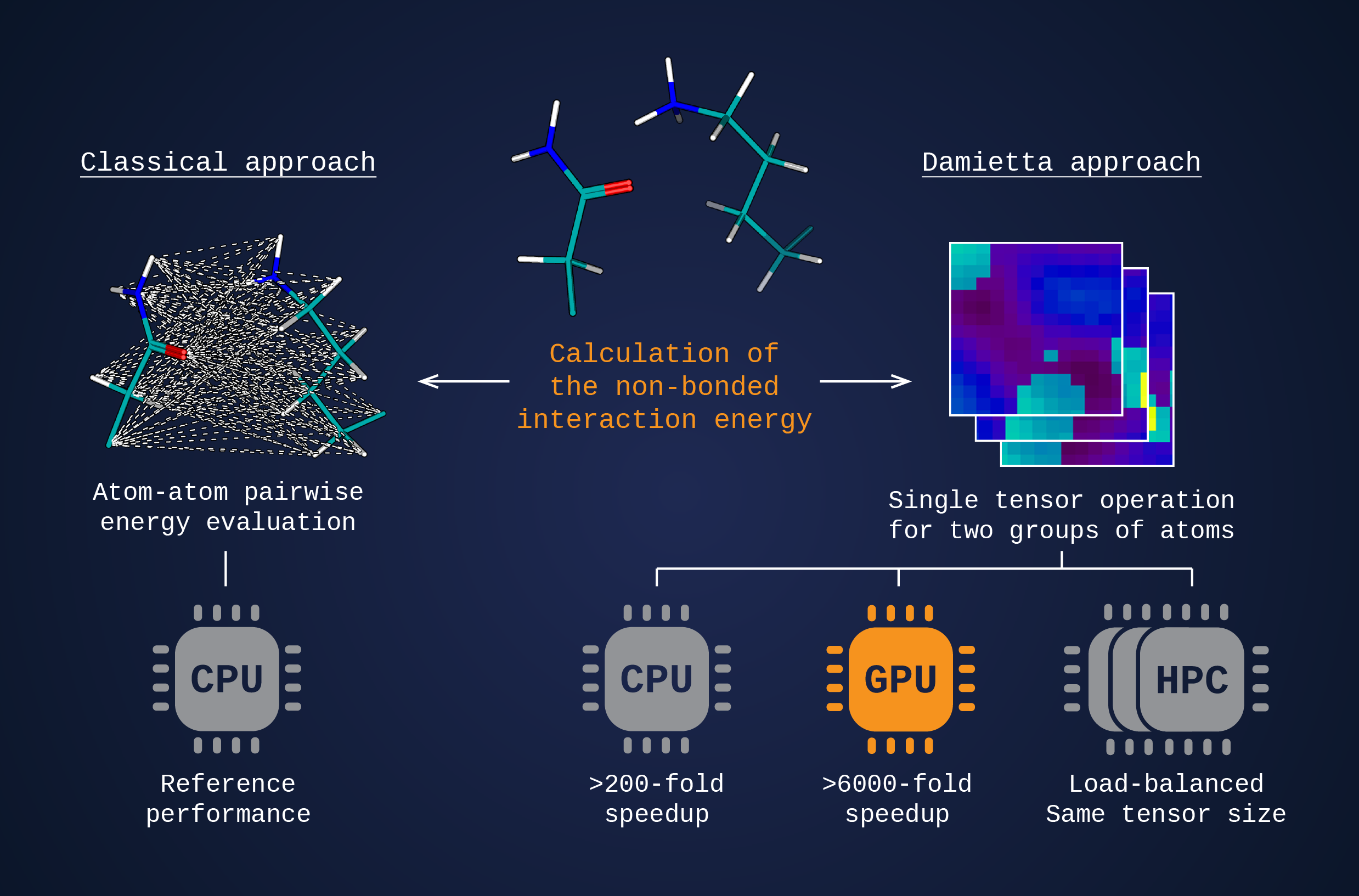
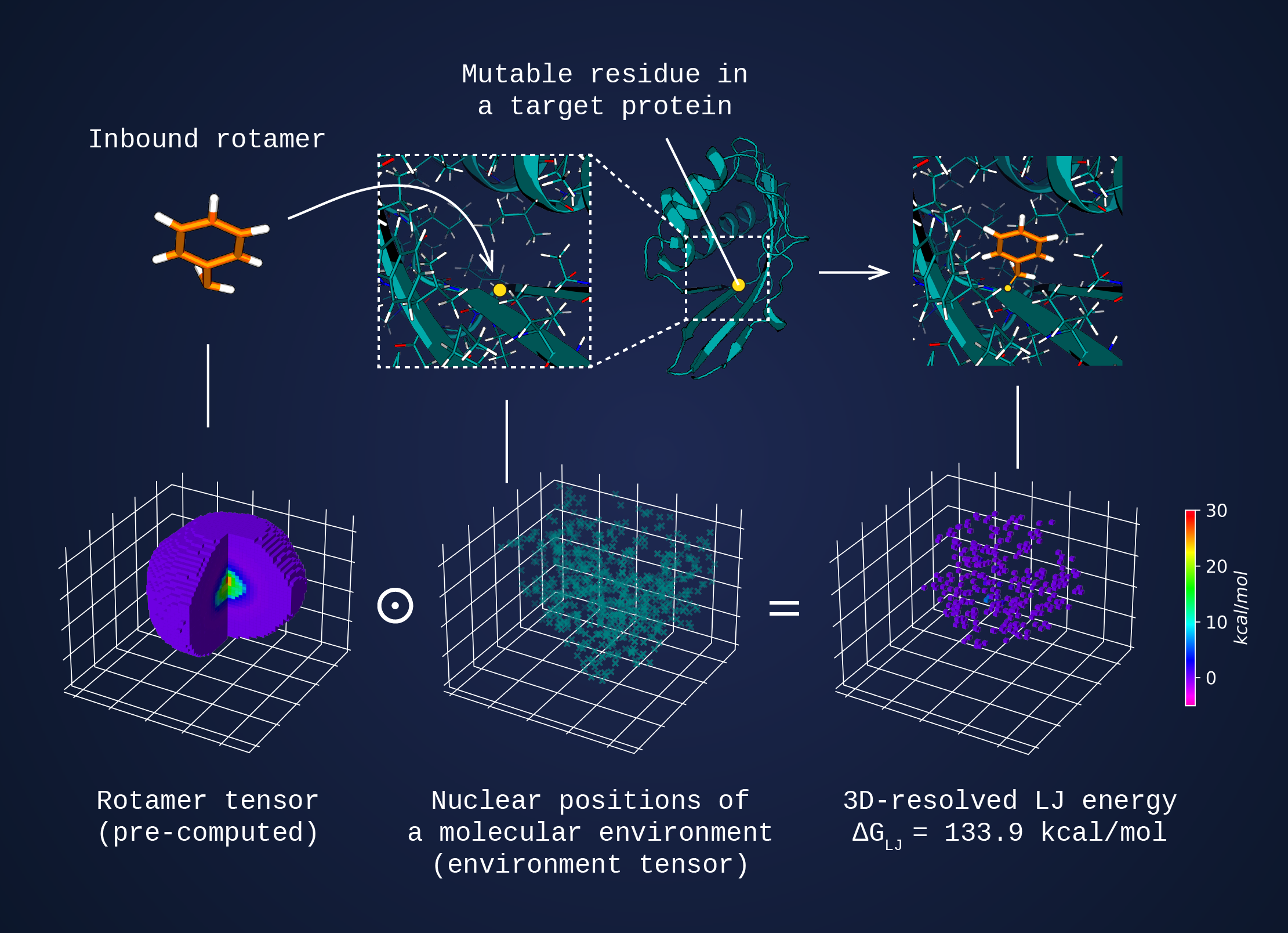
In such a setup, the dense atomic interaction fields for each rotamer are precomputed and stored as 3D projections (rotamer tensors) in a rotamer library. In turn, the chemical environment surrounding the side chain at the designable position is mapped on the fly as a simple 3D histogram of atomic positions and charges (environment tensor). The interaction energy is calculated through multiplying a rotamer tensor by an environment tensor.
The figure below illustrates the calculation of Lennard-Jones energy.
Unbiased rotamer library
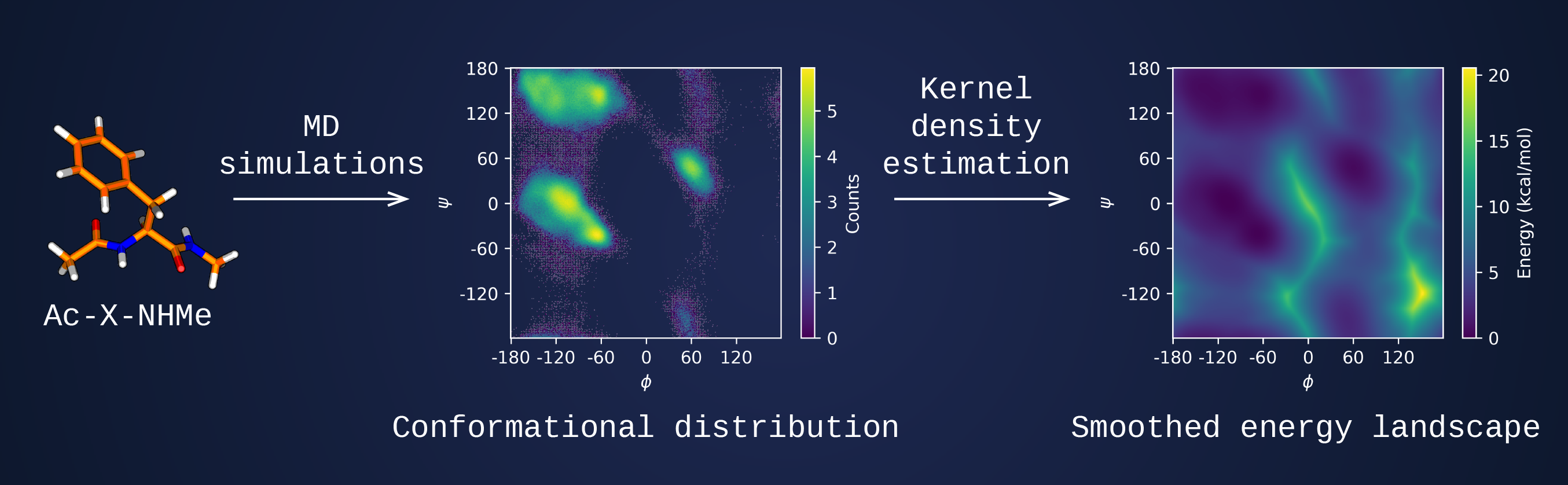
The Damietta design engine relies on the rotamer library generated using molecular dynamics trajectories of capped amino acids. MD-derived ensembles are used to construct the conformational distribution for each amino acid, which in turn gives rise to a probability density function through periodic kernel density estimation. Such library samples a broader conformational space compared to traditional PDB-derived rotamer libraries. Additionally, the library can be extended to cover on-demand rotamers for non-standard amino acids or ligands.
First principles energy function
Damietta design framework relies on an energy function derived from an established molecular mechanics force field (CHARMM). The same force field is used to create the rotamer library. This provides consistency between different scoring terms and eliminates any training procedure for their parametrization. The energy function is composed of 5 additive terms representing residue-wise internal energies (backbone torsions and side chain conformational distributions) and residue-environment interaction energies (Lennard-Jones, solvation, and electrostatic energies).
More details on the tensorized design engine used in the Damietta Toolkit are described in:
Maksymenko et al., The design of functional proteins using tensorized energy calculations, 2023, Cell Reports Methods (doi:10.1016/j.crmeth.2023.100560).
Grin et al., The Damietta Server: a comprehensive protein design toolkit, 2024, Nucleic Acids Research (doi:10.1093/nar/gkae297).