Availability
The Damietta toolkit is available free-of-charge for all users over the following URL: https://damietta.de
Overview
The Damietta toolkit provides a multi-tool framework, allowing the user to conduct several design and modeling processes on their protein structure. The entire framework of the toolkit is organized such that a protein structure is the center of each operation, and it is the main data flowing between different tools. This allows users to forward their structural models across different applications seamlessly.
In the current version, the toolkit provides six distinct
applications, four of which are
native Damietta tools. These are the combinatorial sampler (cs), symmetric design (sd), single-point
mutagenesis (sp), and static energy (se) applications. In
addition, the toolkit offers the HECTOR tool (hec) for selecting potential binding scaffolds with high shape complementarity to the target epitope, and a molecular dynamics-based minimization tool (lm), that is based on the OpenMM library.
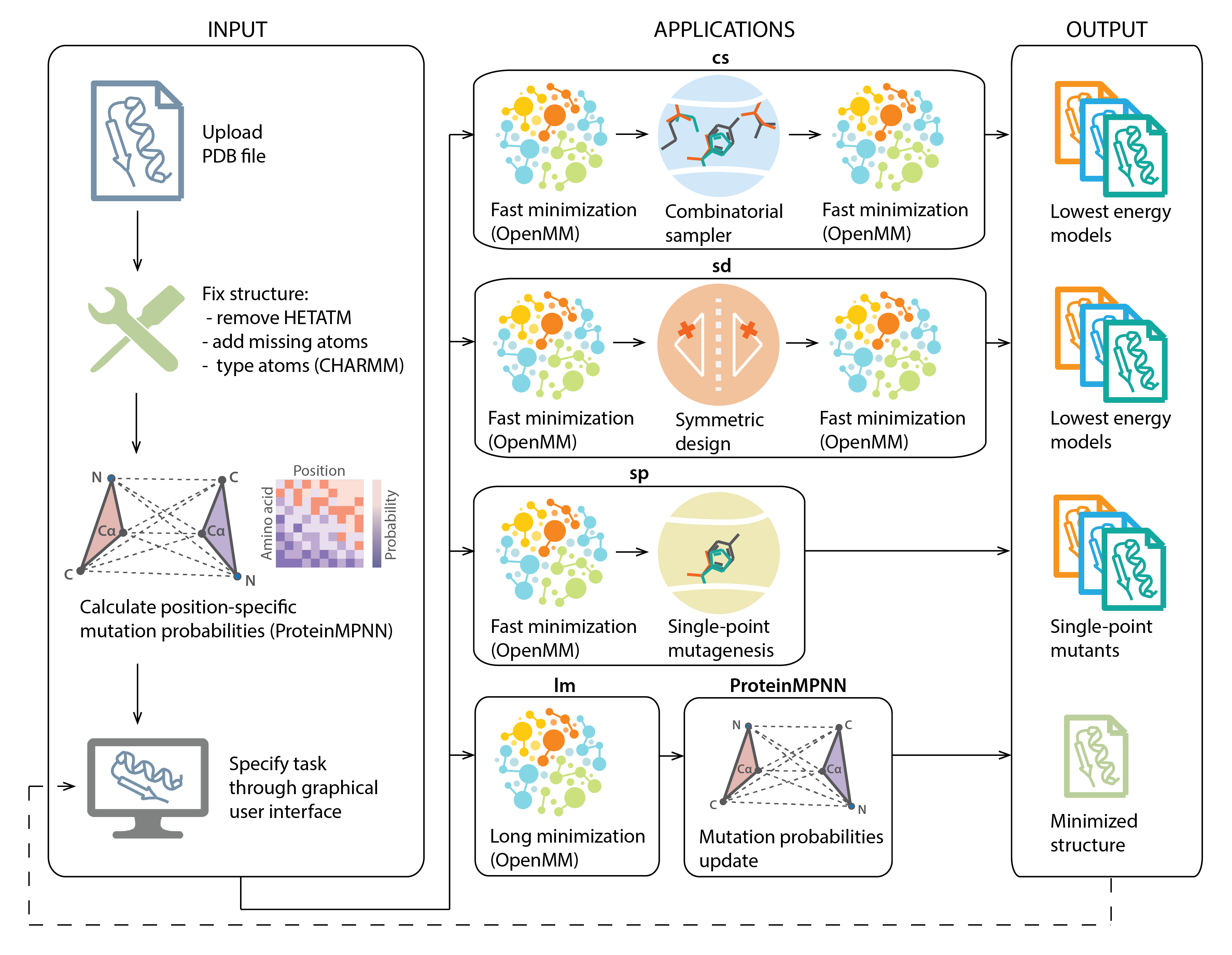
Once the user uploads a PDB file, it is preprocessed, and mutation probabilities at each position are calculated using ProteinMPNN. The protein structure and its sequence are made visually accessible through a molecular viewer. The user at this stage can choose the tool to run, and specify the run parameters. The output is one or more 3D models with their associated scores (e.g., average energy per residue for Damietta applications, or R-factor for the HECTOR tool). The selected output model can be forwarded to any other tool for further operations.
Quickstart guide
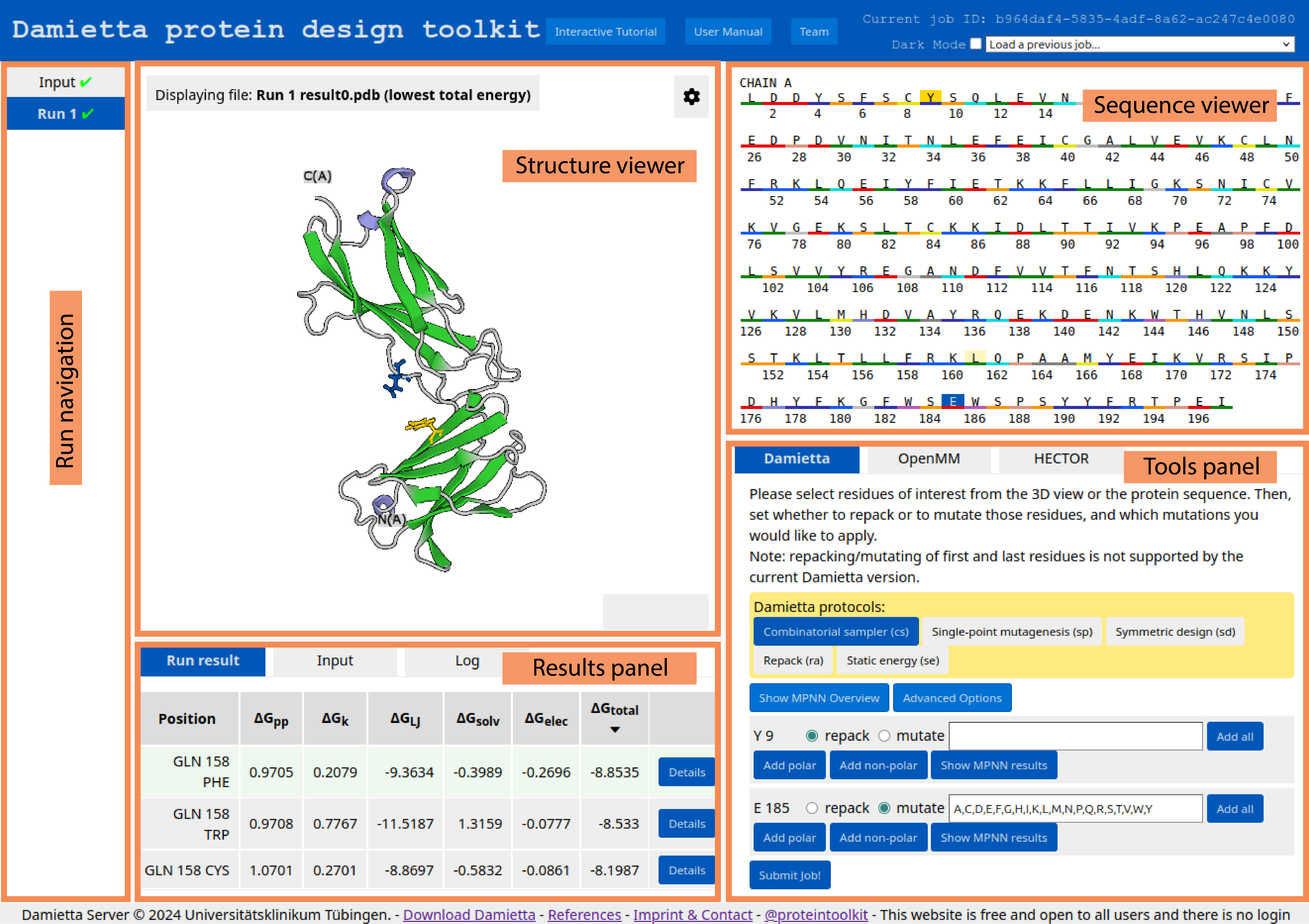
The graphical user interface of the toolkit includes 5 panels. These are run navigation, structure viewer, sequence viewer, tools panel, and results panel. Each of the panel is accessible depending on the current step.
Structure pre-processing. Any workflow starts by uploading a protein structure file in PDB or mmCIF format (https://www.wwpdb.org/documentation/file-format). The upload form is available at the start page:
The server then takes a few seconds to process the structure file by first removing all heteroatoms, such as solvent molecules, ions, and non-proteinogenic ligands or non-standard amino acids that are not yet supported (see Table 1).
| Alanine | ALA | D-Alanine | DAL |
| Cysteine | CYS | D-Cysteine | DCY |
| Aspartate | ASP | D-Aspartate | DAS |
| Glutamate | GLU | D-Glutamate | DGL |
| Phenylalanine | PHE | D-Phenylalanine | DPN |
| Glycine | GLY | ||
| Histidine | HIS | D-Histidine | DHI |
| Isoleucine | ILE | D-Isoleucine | DIL |
| Lysine | LYS | D-Lysine | DLY |
| Leucine | LEU | D-Leucine | DLE |
| Methionine | MET | D-Methionine | MED |
| Asparagine | ASN | D-Asparagine | DSG |
| Proline | PRO | D-Proline | DPR |
| Glutamine | GLN | D-Glutamine | DGN |
| Arginine | ARG | D-Arginine | DAR |
| Serine | SER | D-Serine | DSN |
| Threonine | THR | D-Threonine | DTH |
| Valine | VAL | D-Valine | DVA |
| Tryptophan | TRP | D-Tryptophan | DTR |
| Tyrosine | TYR | D-Tyrosine | DTY |
Then all hydrogens and missing atoms are added in supported amino acids. Afterwards, the atom types of all atoms in the structure are standardized according to the CHARMM36 force field. Finally, all residues are renumbered sequentially, regardless of the input numbering. In case of multi-model PDB, only the first model is processed. As part of the pre-processing, a short structure minimization is also performed.
Upon successful pre-processing, the “Input” job appears in the run navigation panel, and the resulting structure is visualized in the structure viewer:
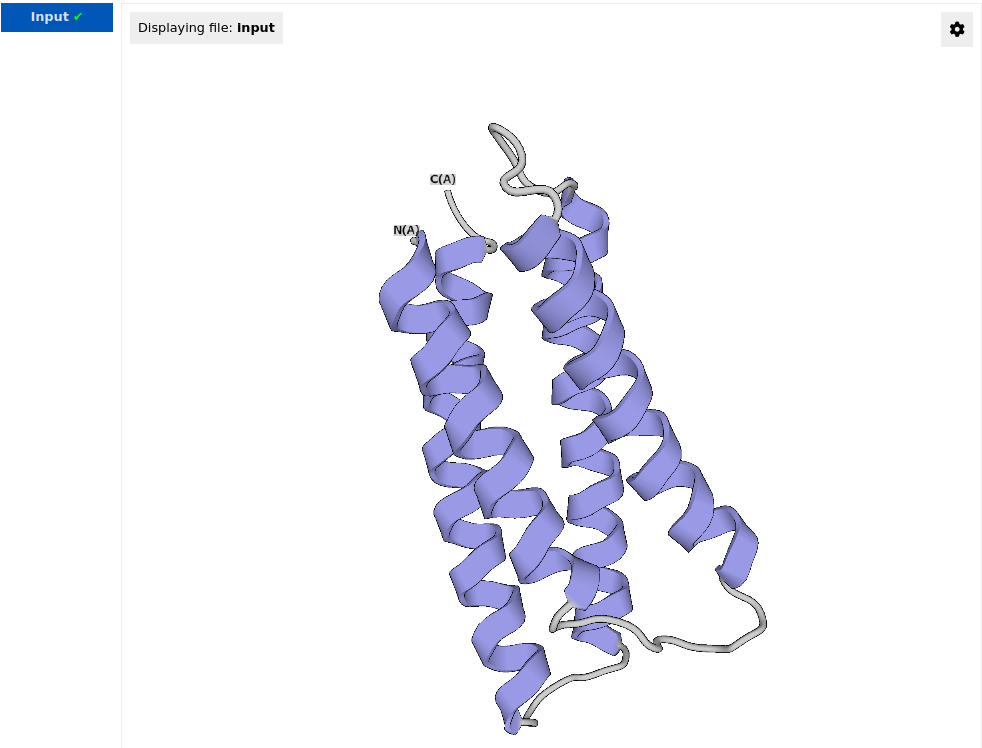
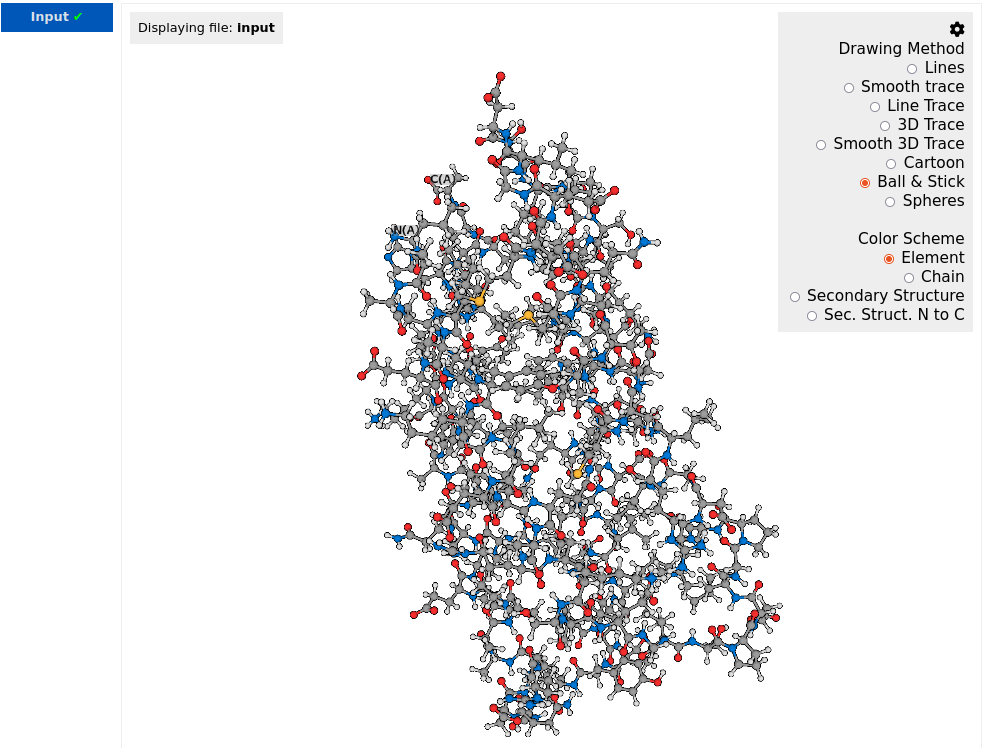
By clicking the settings button ![]() , user can select the drawing method and the color scheme for structure representation. In the example below, the drawing method is set to "Ball & Stick" and the color scheme is set to "Element":
, user can select the drawing method and the color scheme for structure representation. In the example below, the drawing method is set to "Ball & Stick" and the color scheme is set to "Element":
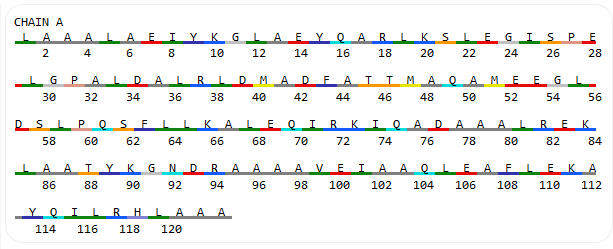
The sequence of the uploaded protein is shown in the sequence viewer:
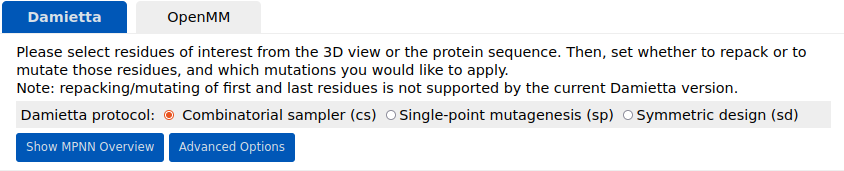
Using the tools panel, the user can choose the tool to run and specify the run parameters. The design, molecular dynamics, and docking routines are available under the "Damietta", "OpenMM", and "HECTOR" tab, respectively:
Combinatorial sampler (cs). The
combinatorial sampler is a powerful tool to introduce a large number of
mutually-compatible mutations or side chain conformations
simultaneously. The actual sampling is performed within a fixed backbone
context, but is preceded and followed by a short spans of structure
minimization to relax the backbone and side chain atoms.
By setting the radio button ![]() to
to
cs tool and choosing the amino acid
positions of interest, a new list of selected positions appears at the
bottom right. The amino acid positions can be selected by clicking on
the desired positions in either the structure or the sequence viewer.
The user can then define the target positions for repacking or
mutagenesis. Repacking a residue changes its conformation to minimize
its energy, typically evaluating 100 unique conformers per a given
backbone conformation. Conversely, mutating a residue evaluates all 100
conformations of the starting residue as well as of all the specified
mutants, and identifies the lowest-energy mutants. The entire routine
combinatorially evaluates and minimizes the energy across all of the
specified residues (via a tree-swarm algorithm), and reports finally up
to 5 unique-sequence designs. Reforwarding the output of the
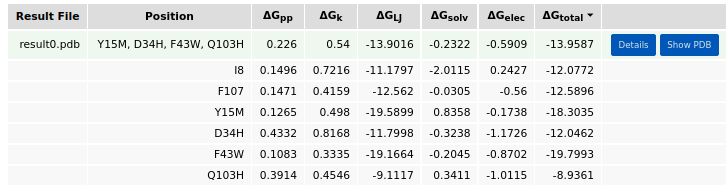
cs tool to itself for further design iterations is generally recommended until the average energy per residue does not improve markedly. In the example below, two residues of the input structure are repacked (I8 and F107), and four positions are mutated (Y15, D34, F43, and Q103). The choice of mutations to sample from is critical, as reducing the number of options would greatly shorten the computational time. Using the associated buttons, the user can select predefined sets of amino acids (e.g., non-polar amino acids for position 15 or polar amino acids for position 34). Alternatively, the user can manually specify amino acids of their choice. For standard amino acids, both three-letter and one-letter codes are supported, whereas non-standard amino acids must be specified using the codes listed in Table 1).
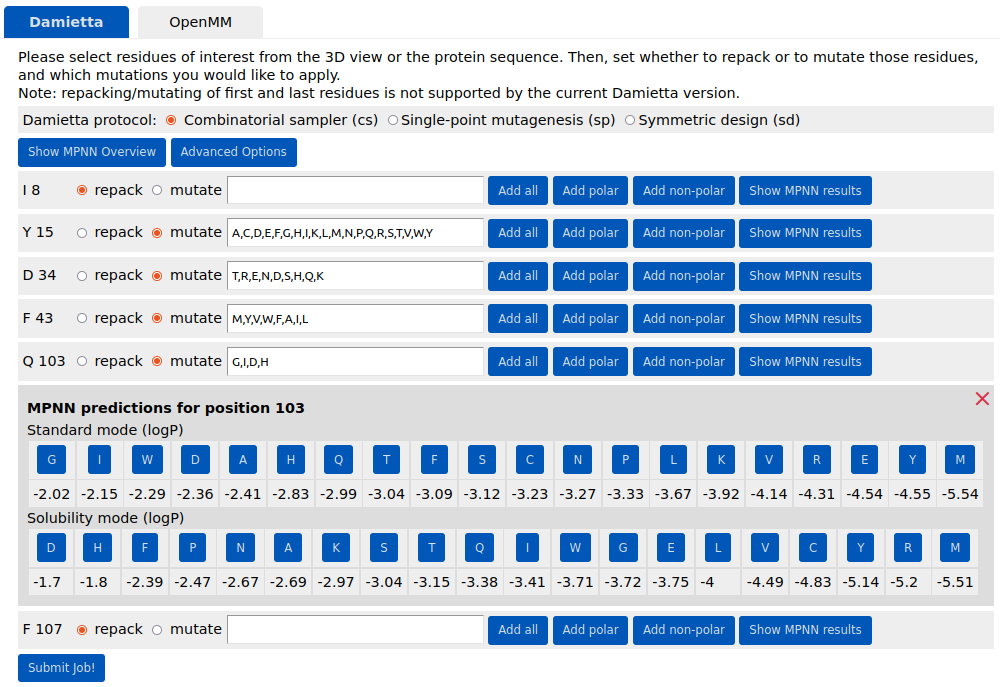
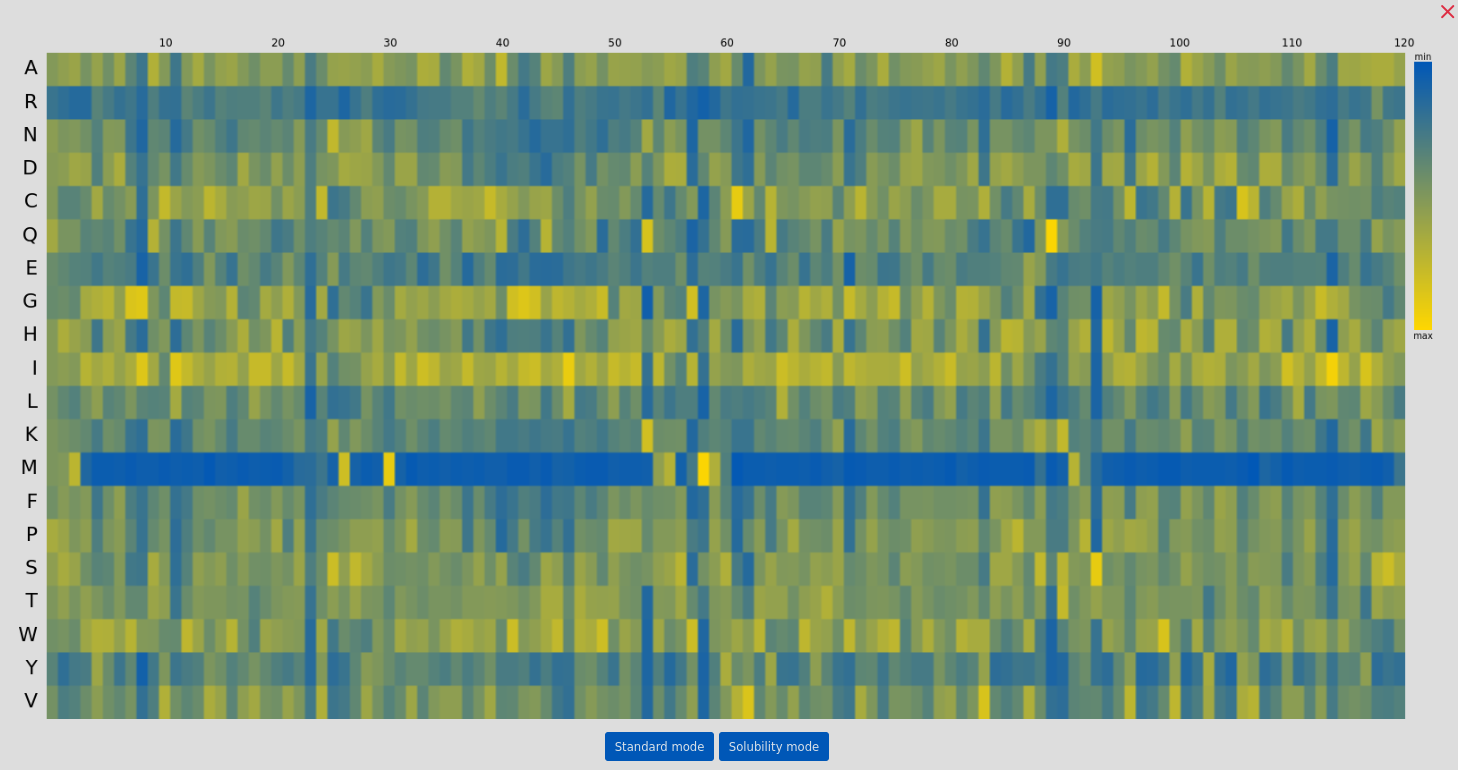
The user can also make guided guesses using the suggestions of ProteinMPNN, by showing the predicted log probabilities and clicking on the highest likely mutations. In the example above, the most likely mutations in the standard mode and the solubility-enhancing predictions mode are selected at position 103. Visualizing the predicted log probabilities is possible by clicking the "Show MPNN Overview" button:
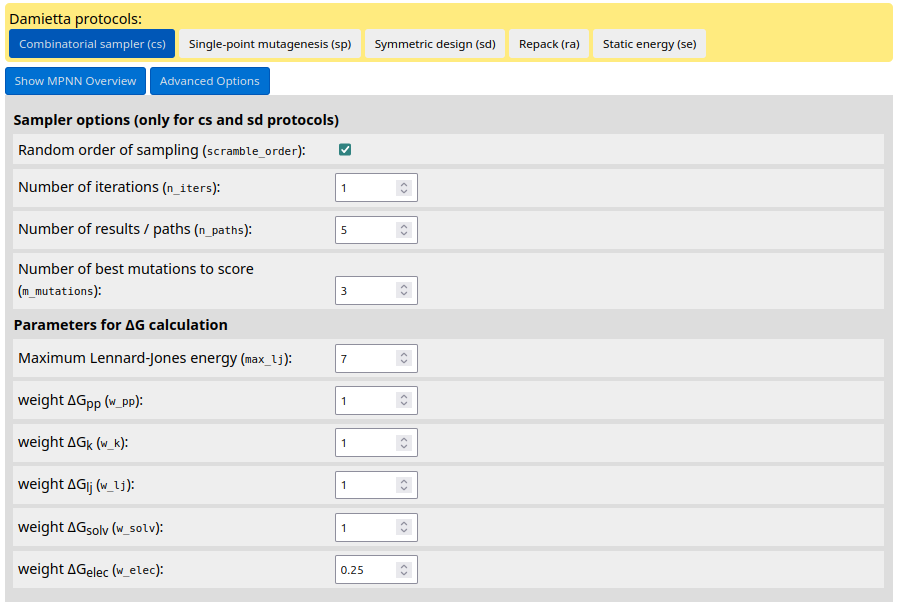
In case of using the Damietta design routines, user can change default sampler options and parameters for energy calculations by clicking the "Advanced Options" button:
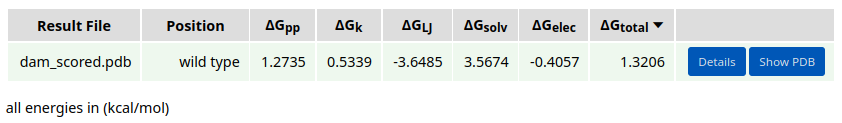
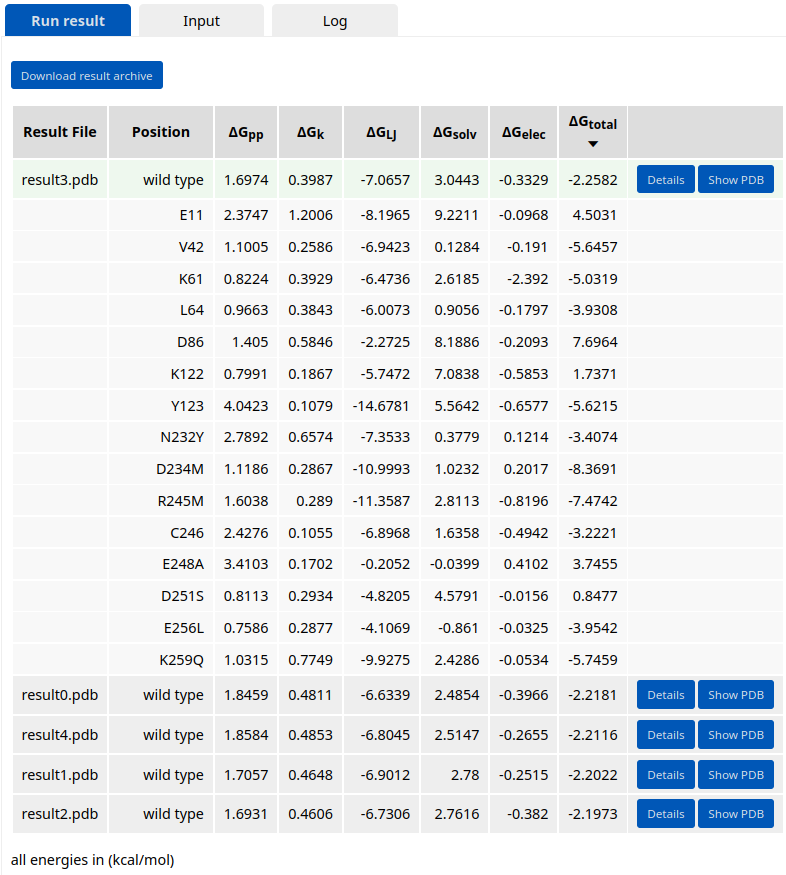
After the job is submitted and completed, the run results will appear in the Results panel. There, the user can swith between Run result, Input, and Log tabs. The resulting design models are sorted by the average energy per residue (\(\Delta\)Gtotal). The broken-down energy terms are also shown. These are the backbone conformation (\(\Delta\)Gpp), side chain conformation (\(\Delta\)Gk), Lennard-Jones interactions (\(\Delta\)GLJ), solvation (\(\Delta\)Gsolv), and electrostatic interactions (\(\Delta\)Gelec) energies.
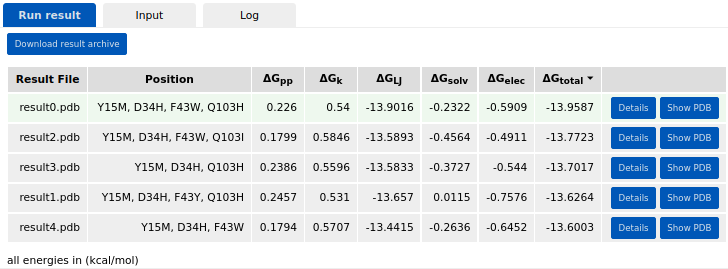
Expanding the details button shows the per-residue energy values. The user can also download the results archive by clicking the respective button. The archive contains a job input in JSON format, a job log, the resulting design models, their FASTA sequences, and a summary table in CSV format. The FASTA file includes 1) the initial input PDB sequence as “wild type” sequence, 2) the last run’s “input” sequence, and 3) the sequences of the resulting designs from the last run. The “Show PDB” button allows the user to visualize and evaluate the introduced mutations in the structure/sequence viewer. The user can further use the selected model for another job (as specified in the tools panel).
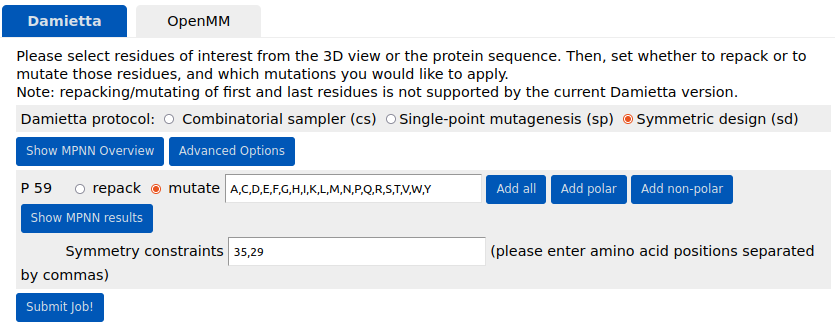
Symmetric design (sd). The
sd protocol performs the combinatorial
sampling operations as cs, while allowing
for introducing sequence symmetry constraints. A mutation at a given mutable position is synchronized with one or more specified positions, which can be provided in an additional field as coma-separated numbers. In the example below, position 59 is mutated to all amino acids, whereby
the same mutations are also evaluated at positions 35 and 29. In this situation, the symmetrically-linked mutations are accepted only if the average energy across all mutable and repackable positions achieved a lower
energy as a result. It is worth noting that the term symmetry here refers only to the imposed sequence symmetry, whereas the sampler minimizes the conformations in an asymmetric manner.
Single-point mutagenesis (sp). The
sp protocol attempts to enforce all the
listed mutations for all the positions individually, and generates the
single-point mutants even if they possess higher energies than the
starting model. However, if the introduced mutation exhibits substantial
steric incompatibility, it will be omitted.
Static energy (se). The
se protocol allows scoring the static structure without repacking. The output structure is identical to the input.
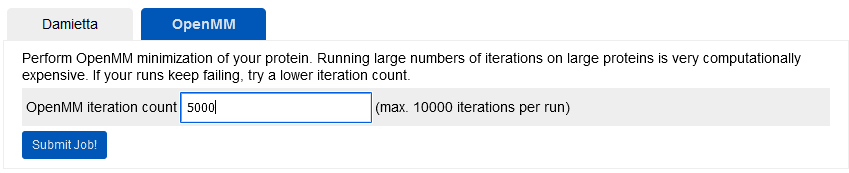
Long minimization (lm). Currently
conjugate gradient minimization is made available under the OpenMM
routines, which is especially useful for relaxing the designed models
after a large number of mutations. The minimized structures can also be
forwarded for further design as needed, and the ProteinMPNN-derived
probabilities will be automatically updated after this long minimization
protocol. The example below specifies 5000 minimization steps for the
selected structure:
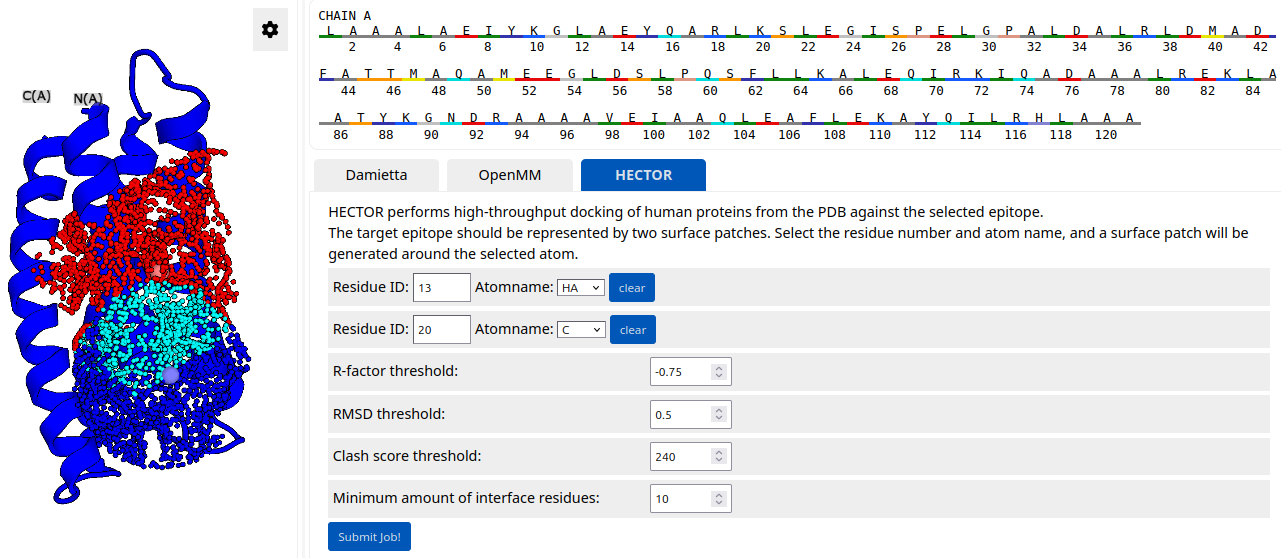
HECTOR (hec). HECTOR is a standalone tool for selecting viable binding scaffolds that can be used as a starting point in a binder design pipeline. It performs ultra-fast complementarity matching to epitopes of interest and outputs the docked scaffolds. The target epitope is described by two surface patches (i.e., query patches). Each surface patch is defined using a cylindrical basis projection, with a radial span of 10 Å and an axial span of 20 Å (for more details, please refer to https://doi.org/10.1002/advs.202502015). First, HECTOR searches the scaffold database, which currently consists of a high-resolution PDB subset of human proteins, and selects scaffolds that contain two subject patches complementary to two query patches. The complementarity metric in this case is the average R-factor between subject and query patches. The lower R-factor is, the higher surface complementarity. The top-ranking scaffolds are then docked against the target structure and filtered based on three docking metrics:
- RMSD of the alignmenet between two query and two subject patches;
- Steric overlap;
- Number of interface residues.
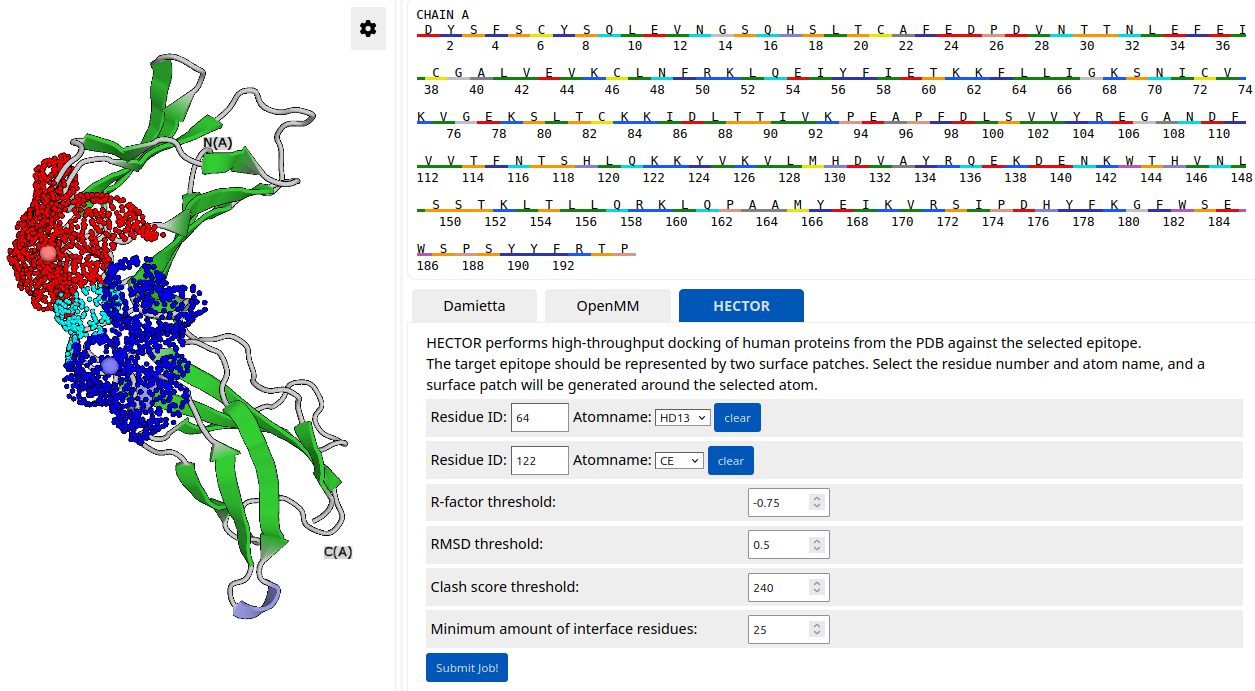
The user can specify a target epitope by selecting two atoms. Two surface patches are then generated around the selected atoms and visualized in the structure viewer (patches shown in red and blue; overlap shown in cyan). In the example below, the target epitope is described by two overlapping surface patches, with their centers located near atom HA of residue 13 and atom C of residue 20, respectively:
HECTOR hits (maximum 50) that pass the filters are listed in the Results panel. The name of a hit corresponds to its PDB ID and chain ID. The user can sort hits by different metrics by clicking on the corresponding table headers. The docked scaffolds can be inspected in the structure viewer using the "Show PDB" button. The results are aslo available for download as an archive. The archive contains a job log, a summary table in CSV format, and PDB files of potential scaffolds docked against the target epitope.
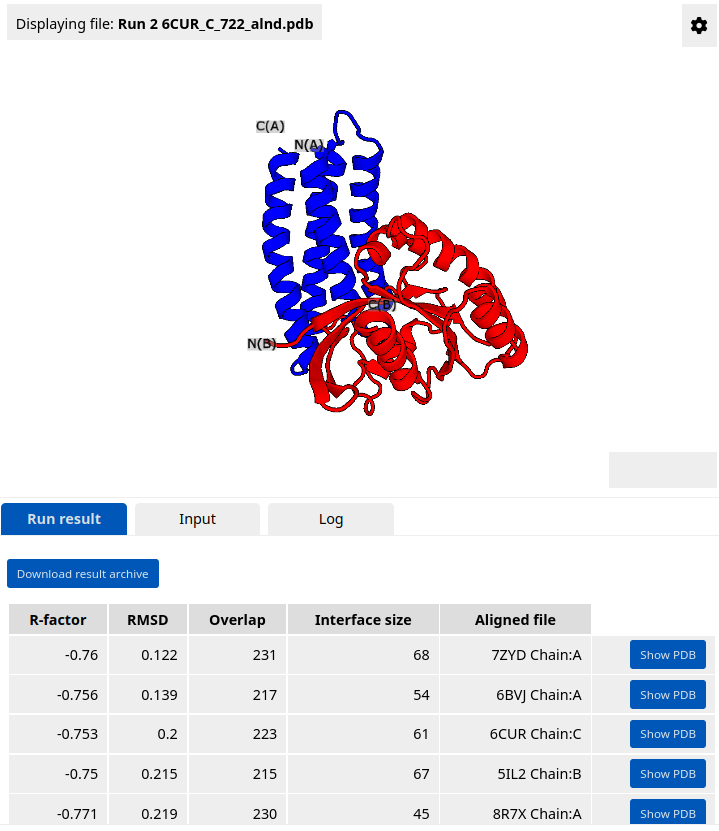
In case no HECTOR hits are found, we recommend slightly loosening the search and filtering parameters compared to the default settings. The user can also try using different minimized models of the input structure to obtain more diverse hits. The resulting docking models can be forwarded to other tools, for example, for minimization with OpenMM or interface design with Damietta.
Case-study: Design of an IL-7Rα binder
This section presents an example of using the Damietta toolkit to design a binder against the ectodomain of the interleukin-7 receptor-α (IL‐7Rα). The aim is to target IL‐7Rα epitope, that overlaps with the native binding site of IL‐7. The 3DI3_B structure from PDB is used as an input structure. On the start page, upload the input file by dragging and dropping it or using the "Browse" button.
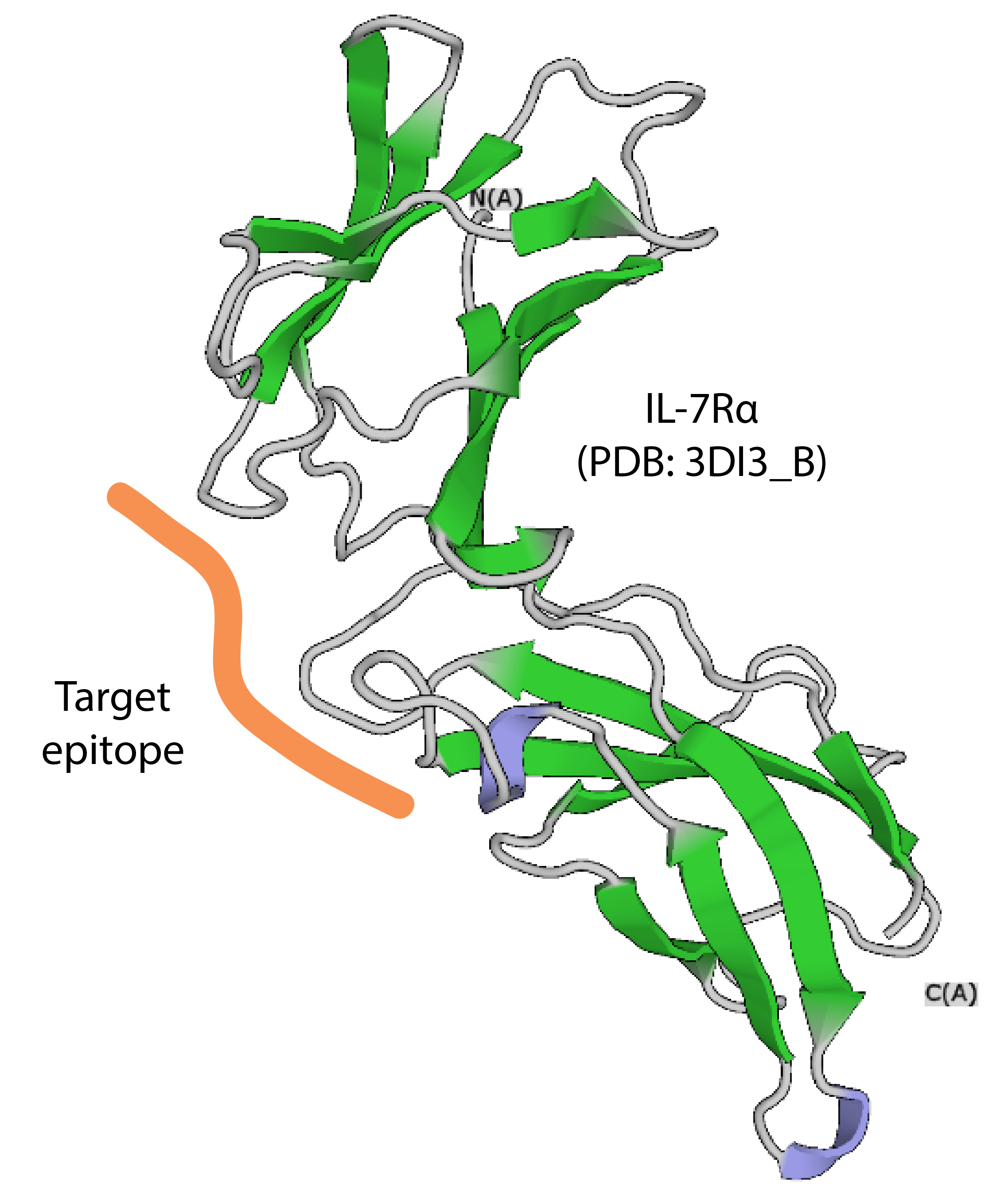
The first step is to identify a suitable binding scaffold that is complementary to the target epitope. This can be done with the HECTOR tool. Under the "HECTOR" tab, specify two residue numbers at the taret epitope (L64 and K122) and select the corresponding atom names from the dropdown menu (HD13 and CE, respectively). Two surface patches are generated around the specified atoms. These surface patches are then used for complementarity matching. For this example, set the following HECTOR thresholds: R-factor < -0.75; RMSD < 0.5; clash score < 240; number of interface residues > 25. Click "Submit job".
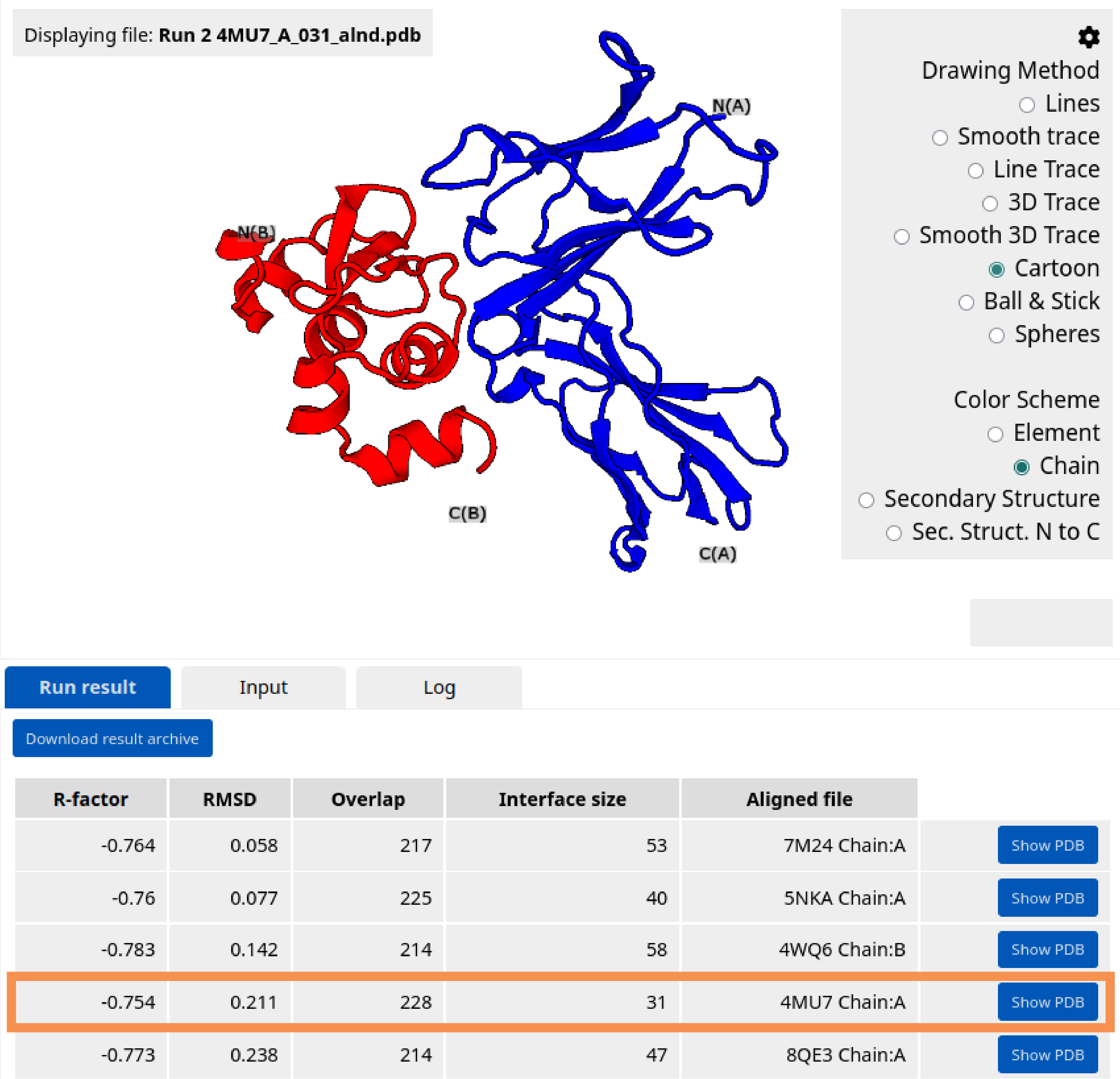
After the HECTOR search is completed, a table of hits appears in the Results panel. Visualize a specific hit using the "Show PDB" button. In the upper-right corner of the structure viewer, set the color scheme to "Chain". The target protein is now colored blue, and the docked scaffold is colored red. Based on the resulting scores and/or visual inspection, the user can choose a hit to proceed. Select 4MU7 Chain:A hit.
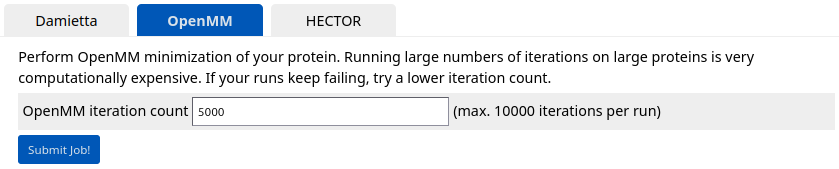
Click "OpenMM" tab to perform minimization of the protein complex. Specify 5000 iterations and submit the job.
Before proceeding to the interface design step, score the energy of the starting complex structure using the se protocol. Click "Evaluate the whole structure".
The static energy of the complex structure is 1.3206 kcal/mol.
The interface design can be performed using cs Damietta application. Specify the following positions for repacking: E11, V42, K61, L64, D86, K122, Y123; and for mutagenesis: N232, D234, R245, C246, E248, D251, E256, K259, W260.
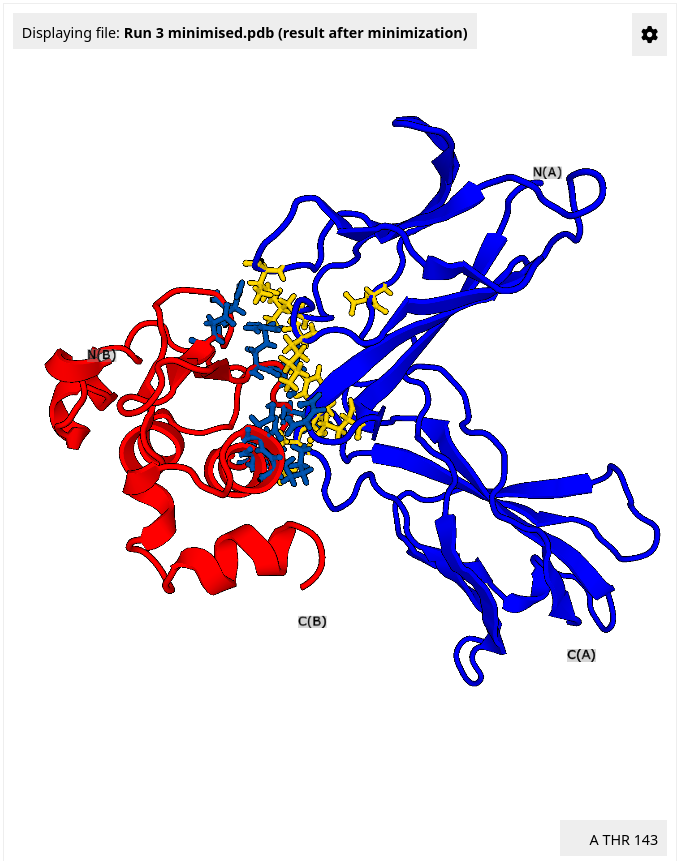
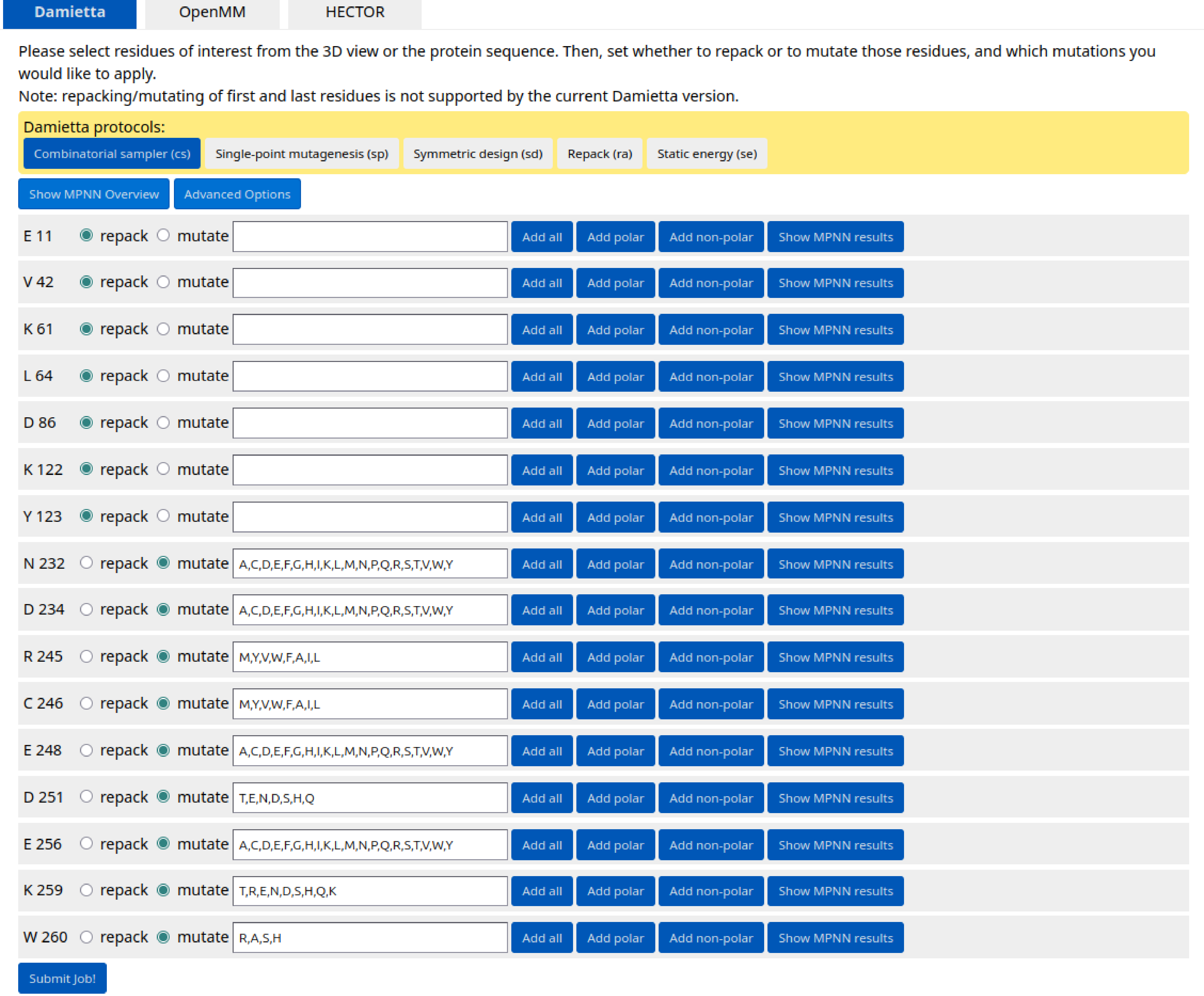
Five top-scored mutants are listed in the results table. Click the "Details" button next to the Result3 design to see which mutations were introduced.
While cs protocol calculates the average enery only for repackable or mutable residues, se application can score the whole structure. Click "Evaluate the whole structure" under the se tab. The static energy of the Result3 design is 0.7489 kcal/mol, which is lower than the energy of the starting template.
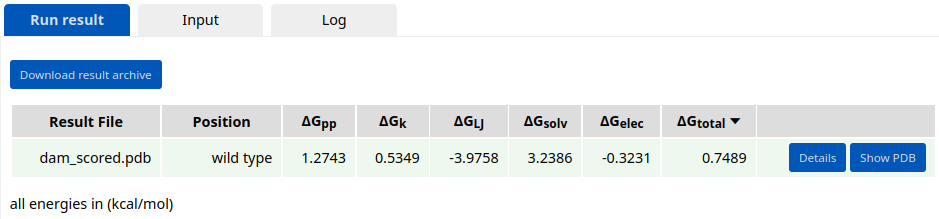
It is recommended to perform several iterations of combinatorial design interlaced with minimization runs until the average energy per residue converges.
Limitations of the Damietta toolkit
The current version of the Damietta toolkit can handle protein structures with maximum 1000 residues. For processing bigger proteins, please contact us or download the full version of the Damietta software: https://bio.mpg.de/damietta
The first (N-terminal) and the last (C-terminal) residues of the protein can not be mutated, since either \(\phi\) or \(\psi\) dihedral angle is not defined for them.
The current version of the Damietta toolkit does not account for any interactions with heteroatoms (e.g. ligands, cofactors, ions, solvent molecules).
References
If you used the Damietta toolkit, please cite:
Grin et al., The Damietta Server: a comprehensive protein design toolkit, 2024, Nucleic Acids Research (https://doi.org/10.1093/nar/gkae297).
If you used one of the Damietta tools, please cite:
Maksymenko et al., The design of functional proteins using tensorized energy calculations, 2023, Cell Reports Methods (https://doi.org/10.1016/j.crmeth.2023.100560).
If you used HECTOR tool, please cite:
Maksymenko et al., A complementarity-based approach to de novo binder design, 2025, Advanced Science (https://doi.org/10.1002/advs.202502015).
If you used OpenMM, please cite:
Eastman et al., OpenMM 7: Rapid development of high performance algorithms for molecular dynamics, 2017, PloS Computational Biology (https://doi.org/10.1371/journal.pcbi.1005659).
If you relied on the ProteinMPNN-provided suggestions, please cite:
Dauparas et al., Robust deep learning–based protein sequence design using ProteinMPNN, 2022, Science (https://doi.org/ 10.1126/science.add2187).